

Миотония, миоплегия и другие наследственные нервно-мышечные заболевания. Нервно-мышечныезаболевания
Миопатии

Миопатии — группа заболеваний, основу которых составляют различные нарушения в метаболизме и строении мышечной ткани, приводящие к снижению силы пораженных мышц и ограничению двигательной активности. Типичными чертами миопатии являются: прогрессирующая мышечная слабость, развитие мышечных атрофий, снижение сухожильных рефлексов и тонуса мышц. Установить диагноз миопатии помогают электрофизиологические исследования, биохимические анализы крови и мочи, результаты молекулярно-генетического и гистохимического анализа образцов, полученных путем биопсии мышц. Лечение предполагает комплексное назначение метаболических препаратов курсами 3 раза в год.

Общие сведения
Миопатии относятся к группе нервно-мышечных заболеваний. Характеризуются дистрофическим поражением мышечной ткани (преимущественно скелетной мускулатуры) с выборочной атрофией отдельных волокон (миофибрилл) при полной функциональной сохранности анимальной нервной системы. Отличаются хроническим неуклонно прогрессирующим течением. Как правило, манифестация клинических проявлений миопатии приходится на детский и юношеский возраст. Большую часть случаев заболевания представляет генетическая патология — это так называемые первичные миопатии. Реже встречаются миопатии приобретенного генеза — вторичные или симптоматические.
Причины миопатий
В основе первичных миопатий лежат генетически детерминированные нарушения в функционировании митохондрий и ионных каналов миофибрилл, в синтезе мышечных белков или ферментов, регулирующих обмен веществ мышечной ткани. Наследование дефектного гена может происходить рецессивно, доминантно и сцеплено с Х-хромосомой. При этом внешние факторы зачастую выступают в роли триггеров, запускающих развитие болезни. Подобными «пусковыми» факторами могут являться разнообразные инфекции (хронический тонзиллит, частые ОРВИ, бактериальная пневмония, сальмонеллез, пиелонефрит и пр.), алиментарная дистрофия, тяжелые травмы (перелом костей таза, политравма, ЧМТ и др.), физическое перенапряжение, интоксикации.
Приобретенные миопатии могут развиваться на фоне эндокринных расстройств (гиперпаратиреоза, болезни Иценко-Кушинга, гиперальдостеронизма), хронических интоксикаций (токсикомании, наркомании, алкоголизма, профессиональных вредностей), мальабсорбции и авитаминозов, тяжелых хронических заболеваний (ХПН, хронической печеночной недостаточности, сердечной недостаточности, ХОБЛ), опухолевых процессов.
Патогенез
Наличие генетически детерминированных или приобретенных дефектов метаболитов, участвующих в обмене веществ и построении мышечных волокон, приводит к возникновению и прогрессированию дегенеративных изменений последних. Развивается атрофия миофибрилл, происходит их замещение жировой и соединительной тканью. Мышцы утрачивают способность к сокращению, что обуславливает мышечную слабость и ограничение возможности выполнять активные движения.
Последние исследования выявили у больных различными формами миопатий нарушения функционирования как центральных (на диэнцефальном уровне), так и периферических отделов вегетативной нервной системы, играющих не последнюю роль в патогенезе заболевания. Именно этим можно объяснить типичное для миопатий преимущественное поражение проксимальных отделов конечностей, имеющих более богатую вегетативную иннервацию.
Классификация
Специалистами в области неврологии разработано несколько классификаций миопатий. Наибольшую популярность среди клиницистов получил этиопатогенетический принцип разделения, согласно которому выделяют наследственные, воспалительные, метаболические, мембранные, паранеопластические и токсические миопатии. Среди наследственных миопатий наиболее распространены 3 вида: ювенильная/юношеская форма Эрба, псевдогипертрофическая форма Дюшена и плече-лопаточно-лицевая форма. Реже встречаются скапулоперонеальная, окулофарингеальная, дистальная и др. формы. Отдельной группой идут врожденные миопатии: болезнь центрального стержня, немалиновая и миотубулярная миопатия, диспропорция типов миофибрилл.
Воспалительные миопатии классифицируются как инфекционные – возникающие вследствие инфекционно-воспалительного поражения мышечной ткани при различных инфекционных процессах: бактериальных (стрептококковая инфекция), вирусных (энтеровирусы, грипп, краснуха, ВИЧ), паразитарных (трихинеллез, токсоплазмоз) и идиопатические — дерматомиозит, миозит с включениями, полимиозит, миопатии при коллагенозах.
Метаболические миопатии подразделяются на связанные с нарушением липидного обмена в мышцах (недостаточность ацетил-КоА-дегидрогеназы, дефицит карнитина), обмена гликогена (болезнь Андерсена, болезнь Помпе, гликогеноз III типа, болезнь Мак-Ардля, дефицит киназы фосфорилазы b, дефицит фосфоглицеромутазы), метаболизма пуринов (дефицит фермента МАДА) и митохондриальные миопатии (дефицит редуктазы, АТФ, цитохрома b, b1).
Симптомы миопатий
Большинство миопатий имеют постепенное начало с появления небольшой мышечной слабости в конечностях, более быстро возникающей усталости от ходьбы и другой физической нагрузки. В течение нескольких лет происходит нарастание слабости, появляются и прогрессируют мышечные атрофии, возникают деформации конечностей. Из-за значительной мышечной слабости пациенты с трудом поднимаются с пола и ходят по лестнице, не могут прыгать и бегать. Для того, чтобы встать со стула, им приходится использовать специальные приемы. Характерен вид больного: крыловидно отстоящие лопатки, опущенные плечи, выпяченный вперед живот и усиленный поясничный лордоз. Наблюдается «утиная» походка — пациент передвигается, раскачиваясь в стороны.
Патологические изменения при миопатиях происходят симметрично в мышцах конечностей и туловища. Как правило, мышечные атрофии наблюдаются в проксимальных отделах рук и ног. В связи с этим мышцы дистальных отделов конечностей могут выглядеть гипертрофированными. Такая миопатическая псевдогипертрофия наиболее заметна в мышцах голеней. Наряду с нарастанием мышечной слабости наблюдается постепенное угасание сухожильных рефлексов и прогрессирующее снижение мышечного тонуса, т. е. развивается и усугубляется периферический вялый паралич. Со временем результатом резкого ограничения активных движений становятся контрактуры суставов.
Миопатии могут сопровождаться поражением мимических мышц, что проявляется невозможностью вытянуть губы трубочкой, свистеть, нахмурить лоб или улыбнуться. Поражение круговой мышцы рта приводит к появлению дизартрии, связанной с затруднением произношения гласных звуков.
Клиника некоторых миопатий включает поражение дыхательной мускулатуры, приводящее к возникновению застойной пневмонии и развитию дыхательной недостаточности. Возможны патологические изменения сердечной мышцы с возникновением кардиомиопатии и сердечной недостаточности, мышц глотки и гортани с развитием дисфагии и миопатического пареза гортани.
Особенности отдельных форм миопатии
Ювенильная миопатия Эрба наследуется аутосомно-рецессивно. Патологические процессы начинают проявляться в возрасте 20-30 лет. В первую очередь они охватывают мышцы тазового пояса и бедер, затем быстро распространяются на другие мышечные группы. Вовлечение лицевой мускулатуры не характерно. Начало миопатии в более молодом возрасте приводит к ранней обездвиженности пациентов. При развитии заболевания в старшем возрасте его течение менее тяжелое: пациенты длительно сохраняют способность передвигаться.
Псевдогипертрофическая миопатия Дюшена наследуется рецессивно сцеплено с полом. Болеют исключительно мальчики. Как правило, манифестирует в течение первых 3-х лет жизни, реже — в период от 5 до 10 лет. Типично начало с атрофических изменений мышц тазового пояса и проксимальных отделов ног, сопровождающихся псевдогипертрофией икроножных мышц. Рано возникают контрактуры и искривление позвоночника (кифоз, сколиоз, гиперлордоз). Может наблюдаться олигофрения. Заболевание протекает с поражением дыхательных мышц и сердца (кардиомиопатия отмечается у 90% больных миопатией Дюшена), что является причиной раннего летального исхода.
Плече-лопаточно-лицевая миопатия Ландузи – Дежерина имеет аутосомно-доминантное наследование. Манифестирует в 10-20 лет с поражения мимических мышц. Постепенно слабость и атрофии охватывают мышцы надплечий, плеч и груди. Мышцы тазового пояса обычно не страдают. Характерно медленное течение с длительной сохранностью работоспособности, без сокращения продолжительности жизни.
Скапулоперонеальная миопатия — аутосомно-доминантное заболевание. Его особенностью является развитие атрофий в мышцах дистальных отделов ног и проксимальных отделов рук, а также наличие легких сенсорных нарушений дистальных отделов как нижних, так и верхних конечностей.
Окулофарингеальная миопатия характеризуется сочетанием поражения глазодвигательных мышц со слабостью мышц языка и глотки. Обычно манифестирует двусторонним птозом, затем присоединяются расстройства глотания. Особенностью этой миопатии является ее позднее начало — на 4-6-ом десятилетии жизни.
Дистальная поздняя миопатия наследуется аутосомно-доминантно. Отличается развитием слабости и атрофий в дистальных отделах конечностей: вначале в стопах и кистях, а затем в голенях и предплечьях. Характерно медленное течение.
Особенности клинических проявлений различных форм врожденных, наследственных и метаболических миопатий описаны в самостоятельных обзорах.
Диагностика
Установить диагноз миопатии неврологу помогают электрофизиологические методы обследования: электронейрография (ЭНГ) и электромиография (ЭМГ). Они позволяют исключить поражение периферического двигательного нейрона и, таким образом, дифференцировать миопатию от инфекционной миелопатии, нарушений спинномозгового кровообращения, миелита и опухолей спинного мозга. Данные ЭМГ говорят о характерных для миопатий изменениях мышечных потенциалов – уменьшении их амплитуды и сокращении длительности. О прогрессирующем процессе свидетельствует наличие большого количества коротких пиков.
Биохимический анализ крови при миопатии показывает повышение содержания альдолазы, КФК, АЛТ, АСТ, ЛДГ и др. ферментов. В биохимическом анализе мочи показательным является увеличение концентрации креатинина. В установлении формы миопатии первостепенное значение имеет биопсия мышц. Морфологическое исследование образцов мышечной ткани выявляет наличие беспорядочно разбросанных атрофированных миофибрилл среди практически сохранных и гипертрофированных мышечных волокон, а также замещение участков мышечной ткани на соединительную или жировую. Постановка окончательного диагноза возможна только после сопоставления результатов гистохимических, иммунобиохимических и молекулярно-генетических исследований.
С целью диагностики поражений сердечной мышцы пациенту с миопатией могут быть назначены консультация кардиолога, ЭКГ, УЗИ сердца; при подозрении на возникновение пневмонии — консультация пульмонолога и рентгенография легких.
Лечение миопатий
В настоящее время патогенетическое лечение миопатий находится в состоянии научных экспериментов в области генной инженерии. В клинической практике применяется симптоматическая терапия, состоящая в основном в улучшении метаболизма мышечной ткани. С этой целью применяют витамины Е, В1, В6, В12, АТФ, неостигмин, аминокислоты (глютаминовую кислоту, гидролизат из мозга свиньи), антихолинэстеразные препараты (амбеноний, галантамин), анаболические стероиды (нандролона деканоат, метандиенон), препараты калия и кальция, тиаминпирофосфат. Комбинации из нескольких препаратов назначают курсом 1-1,5 мес. 3 раза в год.
Медикаментозное лечение миопатий дополняют физиотерапией (электрофорез с неостигмином, ионофорез с кальцием, ультразвук), легким массажем и ЛФК. Проведение ЛФК может осуществляться в бассейне. Комплекс упражнений должен быть подобран таким образом, чтобы избежать перегрузки ослабленной мускулатуры. В некоторых случаях пациенты нуждаются в консультации ортопеда и подборе средств ортопедической коррекции (корсетов, обуви).
Основу лечения приобретенных форм миопатий составляет терапия основного заболевания: коррекция эндокринных нарушений, устранение токсического воздействия и дезинтоксикация организма, ликвидация инфекционного процесса, перевод хронического заболевания в стадию устойчивой ремиссии и т. д.
Прогноз и профилактика
Наиболее неблагоприятны в прогностическом плане наследственные миопатии, проявляющиеся в раннем детском возрасте. В остальном прогноз зависит от формы миопатии, вовлеченности в процесс сердечной и дыхательных мышц. Прогноз вторичных миопатий более благоприятный при условии успешного лечения основного заболевания.
Профилактикой первичных миопатий служит тщательный сбор семейного анамнеза и обязательное консультирование у генетика пар, планирующих беременность. Профилактикой вторичных миопатий является исключение токсических воздействий на организм, своевременное лечение инфекционных и эндокринных заболеваний, коррекция метаболических нарушений.
Нервно-мышечные заболевания
Заболевания периферических нервов
Мононейропатия. Изолированное поражение периферических нервов, например, при сдавлении, травме, нарушении кровоснабжения (поражение vasa vasorum).
Системные заболевания, поражающие нервы, чувствительные к сдавлению, например сахарный диабет, или патологические состояния, вызывающие диффузные нарушения сосудистого русла (васкулиты), способны вызывать мультифокальную нейропатию (или множественную полинейронатию).
Полинейропатия. Одновременное множественные поражение периферических нервов вследствие воспалительных процессов, метаболических нарушений или токсических воздействий. Клинически проявляется диффузным, симметричным поражением периферических нервов. В первую очередь страдают дистальные отделы конечностей, причем нижние конечности поражаются раньше верхних.
Наиболее часто встречаются следующие мононейропатии.
Синдром запястного канала
Компрессия срединного нерва в области запястья при его прохождении через канал может произойти:
- изолированно; например, у пациентов с избыточными физическим нагрузками (связанными с характером трудовой деятельности)
- при заболеваниях, характеризующихся повышенной чувствительностью нервных стволов к внешнему воздействию (сдавлению)
- при сдавлении нервного ствола в области запястного канала гипертрофированными тканями (табл. 1).
Таблица 1. Состояния, ассоциированные с синдромом запястного канала
Локальные деформации вследствие остеоартрита, переломов костей
Клинические проявления синдрома запястного капала:
- боль в кисти или предплечье, особенно ночью или при напряжении
- парез (паралич) и гипотрофия мышц возвышения большого пальца (thenar)
- снижение чувствительности в зоне иннервации срединного нерва (рис. 1)
- парестезии по ходу срединного нерва, которые возникают при постукивании в области запястного канала (симптом Тинеля)
- как правило, двустороннее поражение.
Рис. 1. Распределение зон иннервации срединного, локтевого и лучевого нервов на поверхности плеча и предплечья
Диагноз может быть подтвержден при помощи электрофизиологических исследований. Определение содержания в крови глюкозы, гормонов щитовидной железы, СОЭ, могут помочь в установлении правильного диагноза.
Лечение определяется тяжестью состояния больного. Основные лечебные мероприятия:
- фиксация мышц, особенно ночью, в частично вытянутом состоянии, кисть должна при этом находиться в состоянии разгибания
- мочегонные средства — эффект неясен
- введение кортикостероидов в просвет запястного канала
- хирургическая декомпрессия срединного нерва.
Нейропатия локтевого нерва
Локтевой нерв может быть подвержен сдавлению на различном уровне, однако наиболее часто это случается в области локтевого сустава.
- боли и/или парестезии (покалывающего характера), распространяющиеся от локтя вниз по локтевой поверхности к предплечью
- паралич или слабость внутренних мышц кисти (поражение мышц возвышения большого пальца)
- снижение чувствительности в зоне иннервации локтевого нерва (рис. 1)
- при хроническом поражении формируется когтистая кисть.
Определение скорости проведения импульса при помощи электронейрографического исследования позволяет точно установить локализацию поражения локтевого нерва.
При нетяжелом поражении может быть эффективна фиксация руки на ночь, выпрямленной в локтевом суставе, что обеспечивает уменьшение сдавления нервного ствола. При более тяжелом поражении положительный результат обеспечивает хирургическая декомпрессия или транспозиция локтевого нерва, однако полный регресс неврологической симптоматики наблюдается не всегда. Оперативное вмешательство показано при постоянной травматизации локтевого нерва, которая сопровождается постоянным болевым синдромом и/или прогрессирующими двигательными нарушениями (парез).
Парез лучевого нерва
Сдавление лучевого нерва в верхней части предплечья может привести к острому развитию синдрома «свисающей кисти», при этом иногда наблюдается утрата чувствительности в зоне иннервации лучевого нерва (рис. 1). Как правило, данное поражение является следствием длительного пребывания предплечья в непривычном положении, например при свисающей в неудобном положении руке с поручня кресла при алкогольном опьянении («паралич субботнего вечера»).
Парез плечевого сплетения
Помимо острой травмы плечевого сплетения (например, в результате родовой травмы или дорожно-транспортного происшествия у мотоциклистов) поражение плечевого сплетения может быть обусловлено другими причинами. Поражение верхнего отдела сплетения носит название паралича Эрба, а нижнего – паралича Клюмпке.
Добавочное ребро или гипертрофированная соединительная ткань может быть причиной компрессии плечевого сплетения в области верхней апертуры грудной клетки. На определенном этапе развития неврологии и нейрохирургии имела место гипердиагностика данного состояния и, как следствие, высокая частота необоснованных хирургических вмешательств. На сегодняшний день считается, что оперативное вмешательство показано пациентам с нарастающим парезом внутренних мышц предплечья, выраженной утратой чувствительности (по ходу локтевого нерва) и с диагнозом, подтвержденным электрофизиологическими методами обследования. Визуализация плечевого сплетения при помощи МРТ обычно неэффективна. При рентгенографическом обследовании возможно выявление добавочного ребра, но сдавление нервного ствола фиброзной тканью визуализировать не удается.
Бронхогенная карцинома верхушки легкого может прорастать в нижние корешки плечевого сплетения, вызывая усиливающуюся боль в одноименной руке, дистальный паралич и гипотрофию, а также снижение чувствительности в дерматомах С7, С8 и Th10. Возможен также синдром Горнера вследствие поражения преганглионарных вегетативных волокон. Симптоматика имеет сходство с первичными и метастатическими опухолями.
Диагностические трудности возникают при поражении сплетения у больных с карциномой молочной железы после проведенного курса лучевой терапии, так как неврологический дефицит может быть следствием распространения опухоли или радиационной плексопатии.
Идиопатическая плечевая плексопатия (невралгическая амиотрофия или нейропатия плечевого нерва)
Состояние характеризуется острой болью в плече и предплечье. Очевидных причин этому нет, хотя заболевание может возникнуть после прививки или операции. После регресса болей (через несколько дней или недель) появляется частичный паралич и слабость окололопаточной группы мышц, а также более удаленных мышечных групп верхней конечности. Особенно подвержена поражению передняя лестничная мышца, атрофия которой сопровождается развитием крыловидных лопаток (рис. 2). Поражение, как правило, одностороннее, с минимальными чувствительными нарушениями. Электрофизиологические исследования зачастую малоэффективны, хотя могут выявляться признаки денервации пораженных мышц. Состав СМЖ не изменен. Специфического лечения не существует, у большинства больных через 1,5-2 года наступает спонтанное выздоровление.
Рис. 2. Крыловидные лопатки
Компрессия латерального кожного нерва бедра, проходящего под паховой связкой; характеризуется потерей чувствительности в соответствующей области (рис. 3). Начало заболевания связано, в частности, с изменением веса пациента (увеличение или уменьшение).
Рис. 3. Парестетическая мералгия. Схема распределения нарушений чувствительности при поражении латерального кожного бедренного нерва
Латеральный подколенный паралич
Подколенный нерв подвержен компрессионным повреждениям в области, где он огибает шейку малоберцовой кости. Проявляется синдромом свисающей стопы (вследствие пареза разгибателя стопы). Одновременно появляются слабость при тыльном разгибании и отведении стопы с утратой чувствительности разной степени выраженности. Данное состояние часто встречается у иммобилизованных пациентов и у пациентов с повышенной чувствительностью нервных стволов к сдавлению, например при сахарном диабете. Свисающая стопа может быть следствием поражения поясничного корешка (обычно L5). Следует отличать данный синдром от поражения малоберцового нерва, для которого характерна сохранная внутренняя ротация стопы, так как задняя большеберцовая мышца иннервируется большеберцовым нервом, а не малоберцовым. Однако требуется электрофизиологическое исследование для уточнения локализации поражения нерва. Повреждение малоберцового нерва обычно обратимо, так как вызывается нарушением проводимости (нейрапраксия). Положительный эффект оказывает фиксация стопы лонгетой.
Причины мультифокальной нейропатии (множественный мононеврит):
- злокачественная инфильтрация (карцинома или лимфома)
- васкулит или заболевание соединительной ткани:
- ревматоидный артрит
- системная красная волчанка
- узелковый периартрит
- гранулематоз Вегенера;
- саркоидоз
- диабет
- инфекционные заболевания:
- проказа
- опоясывающий герпес
- ВИЧ
- болезнь Лайма;
- наследственная нейропатия со склонностью к параличам от сдав-ления.
Наиболее частой причиной мультифокальной нейропатии является васкулит с болями, слабостью и гипестезией в зонах иннервации нескольких периферических нервов. Чаще поражаются нижние конечности. Поражения отдельных нервов постепенно накапливаются, проявляясь асимметричным поражение конечностей.
Диффузное поражение периферических нервов может быть разделено на группы с поражением двигательных, чувствительных или смешанных нервов. Существует патофизиологическая классификация полинейропатии, основным критерием которой является преобладание поражения миелиновой оболочки или непосредственно нервного ствола нерва (демнелинизирующая или аксональная нейропатия соответственно). Причины полинейропатии приведены в табл. 2.
Нервно-мышечные заболевания. Классификация, этиология, неврология, наследственные, хронические. Симптомы и лечение у детей, взрослых
Нервно-мышечными заболеваниями (НМЗ) является группа патологий, которые передаются на генетическом уровне от родителей детям. Нарушаются мышечные функции, снижается двигательная активность. Появляются характерные клинические симптомы.
Патологические процессы развиваются на фоне нарушений функций нервно-мышечных соединений, при поражении мышц и спинномозговых нейронов, нервов. Правильно подобранная терапия не поможет полностью вылечить человека, но позволит улучшить качество его жизни.
Этиология и неврология
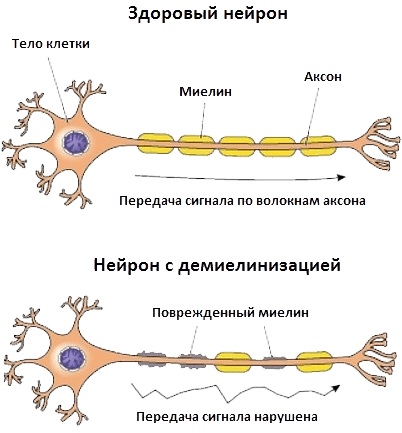
Нервно-мышечные заболевания нарушают нормальную синаптическую передачу импульсов с нервных окончаний к мышечным волокнам. В основе каждого типа патологических изменений лежат аутоиммунные процессы.
Большая группа заболеваний характеризуется не только поражением мышечной ткани, но и периферических нервов, передних рогов спинного мозга. Среди часто диагностируемых патологий выделяют миопатию, миотонию, миастению.
Классификация
Нервно-мышечные заболевания различают по следующим видам:
Описание
Нервно-мышечные заболевания передаются по наследству, чаще появляются у людей, в семье которых были родственники с таким диагнозом.
Приобретенные патологические процессы развиваются в результате гормональных или метаболических нарушений в организме человека. Наблюдается сбой в функционировании иммунной системы. Она вырабатывает клетки, которые атакуют свой организм. Аутоиммунные заболевания приводят к появлению слабости в мышцах.
Нервно-мышечные патологии, сопровождающиеся дистрофическими процессами, поражают следующие области тела человека:
- мышцы;
- нервно-мышечные окончания;
- двигательные нейроны;
- периферические нервы.
При миопатии у человека высоки шансы стать инвалидом в результате утраты подвижности. Все виды нервно-мышечных заболеваний без своевременной терапии влекут за собой последствия. Это может быть не только инвалидность, но и смерть человека.
Стадии и степени
Нервно-мышечные заболевания протекают по стадиям. Определить этап развития патологических процессов поможет врач невролог при помощи медицинской диагностики.
Клиническая картина зависит от скорости развития патологических процессов и степени тяжести заболевания. Установить точный диагноз поможет врач невролог.
Симптомы
Основной признак нервно-мышечных заболеваний – это слабость мускулатуры. Клиническая картина зависит от области поражения (плечевой пояс, бедра, таз, нижние конечности).
В большинстве случаев у пациентов диагностируют следующие симптомы:
- снижается мышечный объем;
- наблюдаются болезненные спазмы;
- непроизвольно сокращаются мышцы;
- пораженные ткани немеют;
- снижаются сухожильные рефлексы;
- больной ощущает покалывание;
- двоится в глазах (диплопия);
- нарушаются глотательные и дыхательные рефлексы.
При нервно-мышечных заболеваниях опускаются веки, мышечная слабость проявляется симметрично и постепенно прогрессирует. В большинстве случаев при развивающейся мышечной дистрофии слабость возникает в области тазового и плечевого пояса. То же самое касается проксимальных отделов конечностей.
Иногда невральная амиотрофия сопровождается парестезией, нарушением глубокой или поверхностной чувствительности. Клинические признаки нервно-мышечных заболеваний проявляются постепенно. По мере прогрессирования патологических процессов человек теряет способности самостоятельно обслуживать себя. То же самое касается передвижения.
Причины появления
Нервно-мышечные заболевания в большинстве случаев возникают по причине аутоиммунных патологий.
Провоцирующим фактором также являются следующие обстоятельства:
- наследственный фактор;
- поражение периферических нервов и мотонейронов спинного мозга;
- сбои в функционировании нервно-мышечных соединений;
- отравление организма различными веществами;
- врожденный сбой метаболизма;
- патологические изменения в мышцах.
Нервно-мышечные заболевания также развиваются на фоне нарушений работы двигательного нейрона в области ствола головного мозга.
Определить причину и поставить точный диагноз поможет врач невролог. Учитывая состояние пациента, степень развития патологических процессов и индивидуальные особенности человеческого организма, специалист подберет эффективное лечение.
Диагностика
Медицинское обследование позволит врачу установить точный диагноз. Тестирование специалист назначает пациенту, учитывая его жалобы и симптоматику.
Для диагностики нервно-мышечных заболеваний назначаются следующие методы обследования:
Источники:
http://www.krasotaimedicina.ru/diseases/zabolevanija_neurology/myopathy
http://medbe.ru/materials/nevrologiya/nervno-myshechnye-zabolevaniya/
http://healthperfect.ru/nervno-myshechnye-zabolevaniya.html

