

Что такое дистрофия стопы. Как реализуется лечебный процесс? Окулофарингеальная форма болезни
Окулофарингеальная мышечная дистрофия

Окулофарингеальная мышечная дистрофия – наследственное заболевание, которое имеет как аутосомно-доминантный, так и аутосомно-рецессивный вариант наследования, характеризуется преимущественным поражением мышц лица и головы. Симптомами этого состояния выступают расстройства глотания (дисфагия), птоз век, слабость мимических мышц и офтальмопарез. Иногда развивается общая мышечная слабость. Диагностика окулофарингеальной мышечной дистрофии производится на основании данных настоящего статуса пациента, электромиографии, молекулярно-генетических исследований. Специфического лечения патологии не существует, применяют паллиативные и симптоматические мероприятия.
Общие сведения
Окулофарингеальная мышечная дистрофия (ОФМД) – генетическая патология, которая проявляется поражением мышц головы и отчасти конечностей и имеет особенный характер обуславливающей ее мутации. Исторически первой была открыта аутосомно-доминантная форма заболевания – в 1962-м году Виктор с командой коллег наблюдал у представителей трех семей необычное сочетание дисфагии с птозом век, симптомы со временем прогрессировали и приводили к слабости поясов верхних и нижних конечностей. Изучение наследственного анамнеза больных доказало аутосомно-доминантный механизм наследования окулофарингеальной мышечной дистрофии. Затем в 1975-м году японские исследователи Сатоёши и Киношита описали сходные симптомы миопатии у девочки, имеющей здоровых родителей – этот факт, а также более раннее развитие проявлений указывало на аутосомно-рецессивную разновидность ОФМД. На сегодняшний день точные цифры встречаемости заболевания не выяснены, отмечено только более частое возникновение в некоторых национальных группах (франкоязычные канадцы, бухарские евреи). По данным современной генетики, окулофарингеальная мышечная дистрофия с одинаковой частотой поражает как мужчин, так и женщин.
Причины ОФМД
Уникальностью окулофарингеальной мышечной дистрофии является характер мутации, обуславливающей развитие этого наследственного заболевания. Дефект выявляется в гене PABPN1, который локализован на 14-й хромосоме. Он кодирует полиаденилат-связывающий белок, относящийся к группе протеинов клеточного ядра, который отвечает за стабилизацию образующихся в ядре молекул матричной РНК за счет их полиаденилирования. В структуре гена присутствует последовательность повторяющихся триплетов GCG, количество их повторов у здорового человека в 98% случаев равно шести. Именно изменение количества повторов данного триплета и обуславливает развитие всех форм окулофарингеальной мышечной дистрофии.
При наличии семи повторов GCG в гене PABPN1, особенно в гомозиготном состоянии, развивается аутосомно-рецессивная форма окулофарингеальной мышечной дистрофии. Однако такое происходит не всегда – примерно у 2% фенотипически здоровых людей наблюдается наличие семи повторов в этом гене, как в гетерозиготном, так и гомозиготном состоянии. Причины, почему у таких лиц не развивается ОФМД, достоверно не изучены. В случае более выраженного нарушения структуры гена PABPN1, при котором наблюдается свыше десяти повторов GCG, возникает аутосомно-доминантная разновидность окулофарингеальной мышечной дистрофии. При этом у гомозигот с таким нарушением (наличием такого количества повторов в обеих аллелях) патология протекает в целом тяжелее и диагностируется в среднем на 15-20 лет раньше.
Таким образом, особенностью окулофарингеальной мышечной дистрофии является тот факт, что это нарушение обусловлено «передозировкой» определенного генетического материала. При этом форма патологии в плане механизма ее наследования и симптомов зависит от масштабов вышеуказанной «передозировки». Наличие генетического дефекта приводит к появлению у полиаденилат-связывающего белка аномального «хвоста» из остатков аминокислоты аланина, что нарушает функции этого протеина. Именно с наличием полиаланиновой последовательности в структуре данного белка связывают появление нитевидных включений в ядрах клеток, которое является одним из признаков окулофарингеальной мышечной дистрофии.
Симптомы ОФМД
Клиническая картина окулофарингеальной мышечной дистрофии зависит от формы заболевания. Наиболее тяжелыми и ранними проявлениями характеризуется аутосомно-рецессивный тип этой патологии – нередко первые симптомы появляются еще в детском возрасте. Сначала регистрируется общая мышечная слабость, пониженная активность мимических мышц, незначительный птоз век. При аутосомно-рецессивной форме окулофарингеальной мышечной дистрофии достаточно быстро развиваются расстройства глотания – дисфагия, которая нередко делает процесс питания попросту невозможным. В дальнейшем возникает офтальмопарез и нарастающая мышечная дистрофия верхних и нижних конечностей.
Аутосомно-доминантный вариант окулофарингеальной мышечной дистрофии характеризуется более доброкачественным течением и поздним возникновением симптомов. Обычно первые признаки заболевания отмечаются в возрасте 40-50-ти лет, изредка появляются в 15-20 лет (у гомозигот по мутантной форме гена PABPN1). Одним из первых симптомов данной патологии является птоз век, к которому вскоре присоединяются дисфагия, снижение активности мимики, слабость других мышц лица и шеи. По мере прогрессирования аутосомно-доминантной окулофарингеальной мышечной дистрофии возникает слабость мышц дистальных отделов конечностей и, в ряде случаев, анального сфинктера, что становится причиной энкопреза. Течение заболевания длительное, по разным данным, может занимать от 5-ти до 15-ти лет.
Диагностика ОФМД
Для диагностики окулофарингеальной мышечной дистрофии применяются методики патогистологического изучения мышечных тканей, электромиографии и изучения наследственного анамнеза больного, а также молекулярно-генетические методы. При осмотре выявляют птоз век, расстройства глотания, общую мышечную слабость, снижение мимической активности. Возраст больных варьируется в зависимости от формы заболевания – при аутосомно-рецессивной разновидности пациентами в основном являются дети, в случае доминантного типа – взрослые 40-50-ти лет (реже 15-25-ти лет). Тщательный анализ наследственного анамнеза позволяет определить механизм наследования окулофарингеальной мышечной дистрофии.
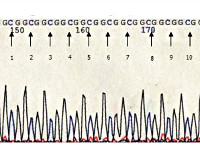
При проведении электромиографии выявляют снижение амплитуды мышечных импульсов, их удлинение и полифазность. Биопсия мышечной ткани с последующим гистологическим изучением обычно обнаруживает наличие в ядрах клеток нитевидных включений, которые иногда приобретают разветвленный характер. При окулофарингеальной мышечной дистрофии нередко возникают нарушения в структуре волокон 1-го типа, при длительно текущем заболевании присоединяются признаки атрофии мышц. Молекулярно-генетическая диагностика этой патологии является надежнейшим и относительно простым (за счет характера мутации) методом определения. С этой целью врач-генетик производит амплификацию участка последовательности GCG с последующим электрофорезом для определения ее размера – увеличение количества повторов GCG указывает на наличие окулофарингеальной мышечной дистрофии. Также возможна пренатальная диагностика этого состояния генетическими методиками, материал для исследования получают методом амниоцентеза или биопсии ворсинок хориона.
Лечение ОФМД
Специфического лечения окулофарингеальной мышечной дистрофии не существует, для уменьшения симптомов заболевания используют различные терапевтические и хирургические мероприятия. Для замедления прогрессирования патологии применяют преднизон, креатин и другие аналогичные препараты, однако их эффективность неодинакова у различных больных. Больным окулофарингеальной мышечной дистрофией назначают разнообразные физические упражнения, которые снижают скорость нарастания мышечной слабости. При выраженном птозе век и связанном с ним нарушении зрения можно использовать специальный скотч для поддержки или же, при относительно сохраненной активности мимических мышц, производить хирургическую коррекцию (блефаропластику).
Наиболее тяжелым осложнением окулофарингеальной мышечной дистрофии, требующим медицинского вмешательства, является дисфагия, в тяжелых случаях приводящая к полной невозможности глотания. В такой ситуации применяют назогастральные или другие зонды для питания больного, иногда накладывают стому. Хирургические методы облегчения симптомов дисфагии (например, рассечение перстнеглоточной мышцы) могут привести к временному облегчению, но в настоящий момент большинство специалистов стараются не использовать такие методики.
Прогноз и профилактика ОФМД
Прогноз окулофарингеальной мышечной дистрофии любого типа (как аутосомно-доминантного, так и рецессивного) неблагоприятный, так как заболевание практически всегда вызывает тяжелые расстройства питания по причине дисфагии. Кроме того, прогрессирующая мышечная слабость конечностей может со временем привести к инвалидизации больного. Из-за нарушений глотания частицы пищи или слюна из ротовой полости часто попадают в дыхательные пути, занося с собой инфекцию и провоцируя достаточно тяжелую аспирационную пневмонию. Именно такое поражение респираторной системы, а также его осложнения чаще всего служат причиной смерти больных окулофарингеальной мышечной дистрофии. Профилактика данного состояния возможна только в рамках медико-генетического консультирования родителей перед зачатием ребенка и пренатальной диагностики в случае наличия такого заболевания у близких родственников.
Что такое дистрофия стопы. Как реализуется лечебный процесс? Окулофарингеальная форма болезни
Окулофарингеальная форма миодистрофии представляет собой миодистрофию позднего возраста, обычно манифестирующую на 6-м десятилетии жизни. В большинстве случаев тип наследования болезни аутосомно-доминантный; описаны единичные семьи с аутосомно-рецессивной передачей окулофарингеальной ПМД [Tome F., Fardeau M., 1994]. Заболевание клинически характеризуется развитием прогрессирующей слабости и атрофии проксимальных отделов конечностей, расстройствами глотания и фонации, птозом, нарушением движений глазных яблок и (на поздней стадии) слабостью лицевой мускулатуры, а патоморфологически-миодистрофическими изменениями указанных групп скелетных мышц и появлением патогномоничных филаментозных внутриядерных включений в мышечных подокнах [Tome F., Fardeau M., 1994; Emery A., 1998].
Ген окулофарингеальной миодистрофии на хромосоме llq 11.2—13 ответственен за синтез ядерного белка РАВP2, служащего фактором полиаденилирования мРНК. У всех больных окулофарингеальной ПМД в мутантном гене обнаруживается увеличение числа копий тринуклеотидных повторов GCG в 1-м экзоне гена: в норме ген содержит 6 тандемных копий повторов GCG, кодирующих полиаланиновый участок в N-терминальной области белка, тогда как у больных число повторов в мутантном гене увеличено до 8-13 копий [Brais B.et al., 1998; Grewal R. etal., 1999;Mirabella M. et al., 2000].
Предполагается, что удлинение полиаланинового участка белка РАВР2 приводит к олигомеризации мутантных белковых молекул, что и лежит в основе образования внутриядерных филаментозных включений. В нормальной популяции у 2% лиц на одной из хромосом обнаруживается «промежуточный» аллель гена, имеющий 7 повторов GCG, наличие которого в гетерозиготном состоянии не сопровождается каким-либо симптомами. Однако у гомозиготных носителей аллеля (GCG) развивается клиника окулофарингеальной ПМД, причем в таких семьях имеет место аутосомно-рецессивный тип наследования болезни [Brais et al., 1998].
Гомозиготность по более длинным GCG-повторам (т.е. двойная доза заведомо мутантного аллеля) характеризуется развитием более тяжелой клинической картины и более ранним дебютом симптомов (в среднем на 18 лет раньше чем у гетерозиготных носителей мутации) [Blumen S. et al., 1999].
Таким образом, с точки зрения мутационного механизма окулофарингеальная ПМД является уникальным заболеванием с двух точек зрения: во-первых, при этой форме миодистрофии имеет место наиболее короткая экспансия повторов по сравнению со всеми другими «тринуклеотидными» заболеваниями; во вторых, различная по тяжести, но одинаковая по характеру мутация в одном и том же гене может служить причиной развития как аутосомно-доминантной, так и аутосомно-рецессивной формы болезни.
Открытие гена РАВР2 и универсального типа мутации в нем позволяет проводить сравнительно простую и достоверную прямую ДНК-диагностику окулофарингеальной мышечной дистрофии, основанную на амплификации тринуклеотидного участка гена с помощью ПЦР. Выявление «промежуточного» ПЦР-продукта с 7 повторами GCG является диагностически значимым в том случае, если оба аллеля имеют такой размер либо второй аллель является заведомо мутантным (>8 повторов GCG).
Окулярная (офтальмоплегическая) и окулофарингеальная форма миодистрофии
Изолированное первичное поражение глазных мыши установлено Говерсом, а затем Мебиусом около 100 лет назад, однако лишь в 1951 г. L. G. Kiloh и в 1974 г. S. Newin подробно описали эту форму миодистрофии. К настоящему времени накопилось достаточно много публикаций, посвященных этой патологии, хотя частота окулярной миодистрофии невелика.
Поражение глазных мышц может быть как изолированным, так и в сочетании с мышцами лица, конечностей, с развитием слабости и атрофии в них. В ряде случаев окулярная миодистрофия может сочетаться с неврологическими и эндокринными проявлениями.
Согласно классификации J. Schmitt, J. Reny (1974), генетически обусловленные прогрессирующие миопатии, поражающие мышцы глаза, делятся на следующие формы.
- Изолированная окулярная миопатия, начинающаяся в молодом возрасте и приводящая к полной наружной офтальмоплегии, обычно без явления диплопии. Диагноз подтверждается данными ЭМГ: на ранних стадиях болезни выявляются изменения, характерные для мышечного уровня поражения. В ряде случаев процесс распространяется на другие поперечнополосатые мышцы, и тогда определяется повышение уровня креатинфосфокиназы, лактатдегидрогеназы и альдолазы в сыворотке крови. Заболевание может быть спорадическим или семейным с аутосомно-доминантным типом передачи.
- Поздняя окулярная миопатия, наблюдающаяся в виде следующих форм:
- окулофарингеальная, при которой, помимо поражения глазодвигательных мышц, обнаруживается слабость мышц глотки и затруднение глотания;
- окулофациальная форма, отличающаяся от формы Ландузи — Дежерина только сохранностью функции круговой мышцы глаза;
- форма, при которой, кроме экстраокулярных мышц, поражаются мышцы проксимальных отделов конечностей;
- окулокардиальная форма, наличие которой оспаривается из-за небольшого числа описанных случаев.
- Окулярная миопатия, сочетающаяся с немышечными поражениями дегенеративного характера, проявляющаяся задержкой развития пирамидной системы, морфологической или функциональной недостаточностью половых желез, сердечной недостаточностью. Это заболевание описывается как синдром Кирнса — Сейра, при котором к офтальмоплегии присоединяется поражение центральной и периферической нервной системы.
Нередко этот синдром обозначается в литературе как окуло-краниосоматическое нервно-мышечное заболевание. Оно обычно выявляется у детей и подростков, у которых слабость экстраокулярных мышц встречается в наборе болезни, не ограничивающейся поражением одних мышц.
У больных часто отмечаются низкий рост, недостаточность умственного развития, атаксия, глухота, пигментная ретинопатия, признаки поражения центрального двигательного нейрона, нередко имеются дефекты проводящей системы сердца. Среди больных этой группы встречаются как спорадические, так и семейные случаи заболевания (описаны аутосомно-доминантный и аутосомно-рецессивный типы).
«Нервно-мышечные болезни»,
Б.М.Гехт, Н.А.Ильина
Источники:
http://www.krasotaimedicina.ru/diseases/genetic/oculopharyngeal-muscular-dystrophy
http://meduniver.com/Medical/Neurology/1503.html
http://www.meddr.ru/nervno-myshechnye_bolezni/nasledstvennye_bolezni_myshc/progressiruuschie_myshechnye_distrofii/14284.html

